
Podcast: Embed
Subscribe: Apple Podcasts | Spotify | Amazon Music | Android | Pandora | iHeartRadio | Blubrry | TuneIn | Deezer | RSS
CardioNerds join Dr. Samid Muhammad Farooqui, Dr. Hiba Hammad, and Dr. Syed Talal Hussain, from the University of Oklahoma Pulmonary and Critical Care Medicine Fellowship Program, in Oklahoma City. The fellows will take us in a fascinating discussion of a case of rapidly progressing dyspnea and pulmonary hypertension in a patient with metastatic breast cancer. They will then reveal an interesting etiology of pulmonary hypertension, where the secret was on the wedge! University of Oklahoma faculty and expert in pulmonary hypertension and right ventricular physiology, Dr. Roberto J. Bernardo provides the E-CPR for this episode. Audio editing by CardioNerds Academy Intern, Dr. Christian Faaborg-Andersen.
A septuagenarian female, with a past medical history of metastatic breast adenocarcinoma, presented to the hospital with worsening dyspnea over a period of 3 weeks. She was found to be in rapidly progressive hypoxic respiratory failure with unremarkable chest x-ray, CTA chest, and V/Q scan. Transthoracic echocardiogram revealed elevated RVSP and a subsequent right heart catheterization showed pre-capillary pulmonary hypertension with a low cardiac index. She was treated for rapidly progressive RV dysfunction with inotropic support and inhaled pulmonary vasodilators until she decided to pursue comfort measures. Wedge cytology came back positive for malignant cells, confirming a diagnosis of Pulmonary Tumoral Thrombotic Microangiopathy (PTTM).
CardioNerds is collaborating with Radcliffe Cardiology and US Cardiology Review journal (USC) for a ‘call for cases’, with the intention to co-publish high impact cardiovascular case reports, subject to double-blind peer review. Case Reports that are accepted in USC journal and published as the version of record (VOR), will also be indexed in Scopus and the Directory of Open Access Journals (DOAJ).
“To study the phenomena of disease without books is to sail an uncharted sea, while to study books without patients is not to go to sea at all.” – Sir William Osler. CardioNerds thank the patients and their loved ones whose stories teach us the Art of Medicine and support our Mission to Democratize Cardiovascular Medicine.

Case Media – When Tumors Take Your Breath Away – University of Oklahoma College of Medicine
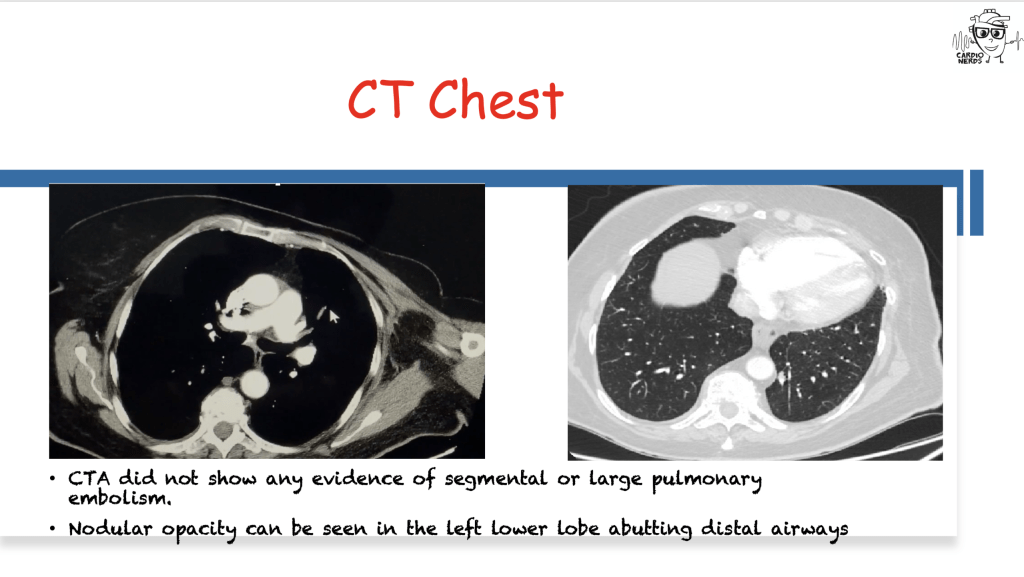






Pearls – When Tumors Take Your Breath Away – University of Oklahoma College of Medicine
- Pulmonary arterial hypertension (PAH) is a progressive disorder of the pulmonary vasculature, characterized by progressive obliteration and remodeling of the pulmonary circulation, resulting in increased pulmonary vascular resistance and increased right ventricular (RV) wall stress, abnormal right ventricular mechanics, and eventually RV dysfunction and death.
- Pulmonary hypertension (PH) is divided into pre-capillary and post-capillary profiles, where pre-capillary PH is hemodynamically characterized by a mean pulmonary artery pressure (mPAP) > 20 mmHg, pulmonary artery wedge pressure (PAWP) ≤ 15 mmHg and a pulmonary vascular resistance (PVR) ≥ 3 Woods Units (WU), and post-capillary PH is defined as mPAP > 20 mmHg, PAWP ≥ 15 mmHg, and PVR can be either < 3 WU (isolated post-capillary PH) or ≥ 3 WU (combined pre- and post-capillary PH). Pulmonary arterial hypertension (PAH) falls under the pre-capillary PH profile.
- Dyspnea on exertion is the most common manifestation of PH, and the most common initial complain. Other symptoms and physical findings such as venous congestion, peripheral edema, signs of RV dysfunction or syncope present later in the disease course. As such, PH has to be considered in the differential diagnosis of dyspnea, especially in cases of undifferentiated or unexplained dyspnea.
- PAH is a chronic but progressive condition, where symptoms progress over the course of months to years. Subacute or rapidly progressive forms of PH (symptoms rapidly worsening over the course of weeks) should warrant consideration for alternative etiologies (i.e., pulmonary embolism or a different cardiopulmonary disorder as the main driver of symptoms), or unique rapidly progressive phenotypes of PAH such as pulmonary tumor thrombotic microangiopathy (PTTM).
- PH in the setting of malignancy warrants special consideration, where the pulmonary vascular disorder could be related to venous thromboembolic disease, external compression of the pulmonary vasculature (if the tumor directly compresses mediastinal structures), related to chemotherapeutic agents (such as tyrosine kinase inhibitors) or thoracic radiotherapy (ie. fibrosing mediastinitis), or related to tumor emboli per se, such as in PTTM. PTTM is a unique manifestation of PH in the setting of malignancy, known to be rapidly progressive, associated with poor RV adaptation, and almost universally fatal. The confirmatory testing of PTTM is by pathology (autopsy), although as in our case, sometimes tumor cells can be identified during cytology of pulmonary artery wedge samples.
Show Notes – When Tumors Take Your Breath Away – University of Oklahoma College of Medicine

1. How do you approach dyspnea?
- Dyspnea is a subjective sensation of uncomfortable breathing. It can be caused by pathologies in cardiac, pulmonary, neuromuscular systems as well as in systemic illnesses. Dyspnea is also a manifestation of psychogenic disorders.
- Presentation of dyspnea can be divided into acute and chronic forms and the etiology can be identified by a thorough evaluation.
- A detailed history and physical exam can help identify the organ system involved. Certain physical signs can be suggestive of the culprit organ system e.g., lower extremity edema in congestive heart failure, increased antero-posterior diameter of the chest in obstructive lung disease, etc. Imaging modalities can be very helpful in determining the cause of dyspnea. Chest radiographs, CT scans of the chest, and echocardiograms can help identify the etiology of dyspnea. Additionally, other testing like pulmonary functions tests can be used too.
2. What are the different Pulmonary Hypertension groups?
Pulmonary Hypertension (PH) is divided into 5 main groups in the WHO classification, as follows:
| Group I | Pulmonary Arterial Hypertension (PAH) | Idiopathic, heritable, drugs, congenital heart disease, liver disease, connective tissue disease, toxins, anorexigens among other causes |
| Group II | PH due to Left Heart Disease | Left sided heart failure, valvular pathology |
| Group III | PH due to Lung Disease | COPD, Interstitial Lung Disease, Sleep Apnea |
| Group IV | PH due to Chronic Thromboembolic Disease | Pulmonary emboli |
| Group V | PH due to Other Causes | Sarcoidosis, ESRD, Sickle Cell Anemia, Chronic Hemolytic Anemia, Certain Metabolic Disorders |
3. How do you approach a patient with Pulmonary Hypertension?
- The goal is to discover an identifiable etiology for proper classification of pulmonary hypertension according to the WHO groups, in order to guide prognostication and management.
- A thorough history and physical exam is the first step in the diagnosis of pulmonary hypertension. Exertional dyspnea is the most common presenting symptom. Due to the nonspecific symptoms, there is often a delay in the diagnosis. Other symptoms include chest pain, fatigue, edema. In severe cases, patients may have syncopal episodes.
- Physical Exam findings concerning for pulmonary hypertension include signs of volume overload (i.e., edema, elevated JVP). Cardiac auscultation may reveal a loud P2 component.
- Laboratory workup includes basic assessment of hematology along with testing for HIV and serological markers of connective tissue diseases. Biomarkers of cardiovascular system like BNP are important in identification and prognostication of pulmonary hypertension.
- Radiological studies like chest radiographs, CT scans of the chest and ventilation/perfusion scans of the lung are used to identify pulmonary pathologies and the presence of thromboembolic disease respectively.
- Echocardiographic assessments are important for diagnosis and assessment of pulmonary hypertension. It allows for the assessment of the left side as well as a detailed analysis of the right side which has diagnostic and prognostic value.
- Finally, the gold standard for diagnosis is a right heart catheterization, which allows for accurate measurements of the pressure in the different chambers of the heart and allows for the phenotyping of pulmonary hypertension.
4. What are the considerations for Pulmonary Hypertension etiologies in patient with malignancy? How is Pulmonary Tumoral Thrombotic Microangiopathy diagnosed?
- Pulmonary hypertension in a patient with malignancy requires special attention.
- Apart from the common reasons for pulmonary hypertension, use of chemotherapeutic agents has been associated with the development of pulmonary arterial hypertension, particularly with Tyrosine Kinase Inhibitors.
- Pulmonary Veno-Occlusive Disease (PVOD) can be precipitated by the use of many chemotherapeutic agents especially alkylating agents.
- Detrimental effects of chemotherapeutic agents on myocytes can cause Group II pulmonary hypertension.
- Chemotherapy and radiation therapy induced lung damage can also cause Group III pulmonary hypertension.
- Large tumors may directly compress mediastinal structures causing elevated pulmonary pressures due to external compression.
- In patients with adenocarcinoma, tumoral thrombotic microangiopathy can result in sub-acute pulmonary hypertension known as Pulmonary Tumoral Thrombotic Microangiopathy (PTTM).
- PTTM results in rapid clinical deterioration and hence requires a high suspicion of index. It is mostly diagnosed postmortem, but can be diagnosed by performing wedge cytology.
5. What is the prognosis of PTTM and how is it treated?
- PTTM carries a grave prognosis. It causes accelerated occlusion of pulmonary arteries resulting in acute to subacute pulmonary hypertension and ensuing RV dysfunction and failure.
- The mainstay of treatment relies on pulmonary vasodilation and slowing the growth of malignant cells.
- Pulmonary vasodilators, especially endothelin receptor antagonists, have been reported to be used.
- Imatinib, a tyrosine kinase inhibitor, has been reported to be used with some improvement in survival.
References –
- Vonk Noordegraaf A, Chin KM, Haddad F, et al. Pathophysiology of the right ventricle and of the pulmonary circulation in pulmonary hypertension: an update. Eur Respir J. Jan 2019;53(1):1801900. doi:10.1183/13993003.01900-2018. Link:
- Bernardo RJ, Haddad F, Couture EJ, et al. Mechanics of right ventricular dysfunction in pulmonary arterial hypertension and heart failure with preserved ejection fraction. Cardiovasc Diagn Ther. Oct 2020;10(5):1580-1603. doi:10.21037/cdt-20-479.
- Simonneau G, Montani D, Celermajer DS, et al. Haemodynamic definitions and updated clinical classification of pulmonary hypertension. The European respiratory journal. 2019;53(1):1801913-1801913. doi:10.1183/13993003.01913-2018.
- Dumitrescu D, Sitbon O, Weatherald J, Howard LS. Exertional dyspnoea in pulmonary arterial hypertension. Eur Respir Rev. Sep 30 2017;26(145)doi:10.1183/16000617.0039-2017.
- Buser M, Felizeter-Kessler M, Lenggenhager D, Maeder MT. Rapidly progressive pulmonary hypertension in a patient with pulmonary tumor thrombotic microangiopathy. Am J Respir Crit Care Med. Mar 15 2015;191(6):711-2. doi:10.1164/rccm.201501-0004IM.
- Price LC, Wells AU, Wort SJ. Pulmonary tumour thrombotic microangiopathy. Lippincott Williams and Wilkins; 2016. p. 421-428.
- Price LC, Seckl MJ, Dorfmüller P, Wort SJ. Tumoral pulmonary hypertension. European Respiratory Review. 2019;28(151)doi:10.1183/16000617.0065-2018.
- Shah AT, Bernardo RJ, Berry GJ, Kudelko K, Wakelee HA. Two Cases of Pulmonary Tumor Thrombotic Microangiopathy Associated with ROS1-Rearranged Non-Small-Cell Lung Cancer. Clin Lung Cancer. Mar 2021;22(2):e153-e156. doi:10.1016/j.cllc.2020.09.020.
- Godbole RH, Saggar R, Kamangar N. Pulmonary tumor thrombotic microangiopathy: a systematic review. Pulm Circ. Apr-Jun 2019;9(2):2045894019851000. doi:10.1177/2045894019851000



